Felipe Ferrão
Written by Guilherme da Silva Pereira, updated by Rodrigo Rampazo Amadeu.
This material is based upon the following TASSEL tutorials: Tassel Pipeline GBS, TASSEL 3.0 / 4.0 Pipeline Command Line Interface, and TASSEL 3.0 UNEAK Pipeline. We apologize, but this post is on Portuguese language. However, you can easily do it by following the command lines inside the chunks.
Este tutorial foi inicialmente desenvolvido para um workshop.
- Introdução
- Antes de começar
- Pipeline TASSEL-GBS
- 1º passo:
FastqToTagCount - 2º passo:
MergeMultipleTagCountPlugin - 3º passo:
TagCountToFastqPlugin - 4º passo:
bowtie2-build - 5º passo:
bowtie2 - 6º passo:
SAMConverterPlugin - 7º passo:
FastqToTBTPlugin - 8º passo:
MergeTagsByTaxaFilesPlugin - 9º passo:
DiscoverySNPCallerPlugin - 10º passo:
MergeDuplicateSNPsPlugin - 11º passo:
MergeIdenticalTaxaPlugin - 12º passo:
GBSHapMapFiltersPlugin
- 1º passo:
- Extra: R/qtl
- Pipeline UNEAK
- Considerações finais
Introdução
Este é um breve tutorial que exemplifica o trabalho de bioinformática de identificação de SNPs a partir dos dados brutos de leituras curtas oriundas da aplicação da técnica de genotipagem por sequenciamento (do inglês, genotyping-by-sequencing – GBS).
Inicialmente, há uma breve explicação sobre a origem dos dados e dos programas utilizados. Em seguida, partimos para a prática em si utilizando o pipeline Trait Analysis by aSSociation, Evolution and Linkage (TASSEL)-GBS de alinhamento e de descoberta da SNPs a partir de dados de GBS. Finalmente, uma pequena demonstração da utilização dos dados obtidos para estimar um mapa genético da população será realizada. Nesta atualização da aula de 28-07-2014 realizada durante o Workshop, foram incluídos os trabalhos com (i) NPUTE, para imputação dos dados perdidos, (ii) arquivo do tipo VCF e (iii) UNEAK, para chamada de SNPs sem genoma de referência.
A utilização desse material está condicionada ao favor de me escrever caso encontre algum erro ou tenha sugestões/críticas – ficarei grato com o feedback.
Antes de começar
Como o pipeline TASSEL requer uma série de pastas, arquivos e programas para funcionar. Então, criar pastas e obter arquivos e programas são as primeiras coisas que devemos fazer.
Pastas de trabalho
Em cada computador, uma pasta denominada gbs-tassel-arroz, previamente
criada na /home do seu usuário linux, já contém todas essas pastas. No
terminal, acesse a pasta e liste seu conteúdo. Assim:
cd gbs-tassel-arroz
ls -l
Para criar as pastas aí dentro, simplesmente rodamos (mas você não precisa fazer isto agora, porque, afinal, já estão criadas):
mkdir fastq reference tagCounts mergedTagCounts alignment topm tbt mergedTBT hapmap hapmap/raw hapmap/mergedSNPs hapmap/mergedTaxa hapmap/filt hapmap/bpec vcf vcf/raw vcf/mergedSNPs vcf/mergedTaxa
Dados
Os dados brutos de leituras curtas que utilizaremos aqui resultaram da aplicação da técnica de GBS em uma população de mapeamento de 176 linhagens endogâmicas recombinantes (do inglês, recombinant inbred lines – RILs) de arroz, Oryza sativa. Elas foram desenvolvidas via técnica de descendência de única semente (do inglês, single seed descent – SSD) do cruzamento entre IR64 (indica) × Azucena (japonica). Os detalhes sobre a população e as estratégias de GBS aplicadas, assim como todos os resultados, podem ser consultados no artigo científico que tornou os resultados (e os dados) públicos. Aqui, por questões didáticas, utilizaremos apenas o output da primeira rodada de GBS realizada (384-plex), mas as análises podem ser estendidas, obviamente, a todos os dados disponibilizados.
Na pasta denominada gbs-tassel-arroz, já foram baixados os arquivos
pertinentes para esta prática contidos no
site. Estes arquivos incluem, além
dos dados brutos de leituras, o key file, contendo as informações
indispensáveis sobre as amostras (como flowcell, lane, sample ID e
barcodes). Para obtê-los, via terminal, utilizamos:
wget http://www.ricediversity.org/data/sets/IR64xAzucena/D0DTNACXX_1-IR64-Azucena.fq.bz2
wget http://www.ricediversity.org/data/sets/IR64xAzucena/D0DTNACXX_1-key-file.tsv
Como o pipeline TASSEL requer um certo estilo para entrada dos
dados brutos, renomeamos o arquivo contendo esses dados. Em seguida, o
arquivo com extensão .bz2 foi descompactado e seu resultado, com
extensão .txt, foi movido para a pasta fastq, como requerido:
mv D0DTNACXX_1-IR64-Azucena.fq.bz2 D0DTNACXX_1_fastq.txt.bz2
bunzip2 -k D0DTNACXX_1_fastq.txt.bz2
mv "D0DTNACXX_1_fastq.txt" ./fastq/
Genoma de referência
Também por demandar um certo tempo em download e descompactação, já disponibilizamos a última versão do genoma do arroz na sua pasta de trabalho. Esse genoma, assim como o de várias outras espécies vegetais, podem ser encontrados no Phytozome (divirtam-se navegando por ele!). Para obter o arquivo em formato fasta do genoma, via terminal, utilizamos:
wget ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Osativa/assembly/Osativa_204.fa.gz
Certa formatação adicional para o programa bowtie2 (o alinhador que
utilizaremos) faz-se necessária. Basicamente, descompactamos o arquivo
de extensão .gz e substituímos “>Chr” por “>chr” e números
sequenciais apenas para designar cada um dos cromossomos. Assim:
gunzip -c Osativa_204.fa.gz > ./reference/Osativa_204.fa
grep '>' ./reference/Osativa_204.fa
sed -i 's/>Chr/>chr/g' ./reference/Osativa_204.fa
sed -i 's/>chrUn/>chr13/g' ./reference/Osativa_204.fa
sed -i 's/>chrSy/>chr14/g' ./reference/Osativa_204.fa
grep '>' ./reference/Osativa_204.fa
Como o arroz tem 12 cromossomos, utilizaremos apenas as primeiras 12 sequências nas análises posteriores.
Programas
O programa de alinhamento de sequências curtas contra um genoma de referência que utilizaremos aqui será o bowtie2 (mas o TASSEL também funciona com o alinhamento via BWA – fica a gosto do freguês!). Há uma publicação sobre e um site mantido pelos desenvolvedores. Fique à vontade para navegar nesses sites enquanto espera suas análises “rodarem”!
Já o TASSEL-GBS é trabalho apresentado neste artigo e todo o material está disponível no site do grupo. Este presente tutorial baseou-se, inclusive, neste material organizado e mantido pelos desenvolvedores. Consultem-no! Há inúmeros e importantes detalhes que omitimos aqui por brevidade.
Neste tutorial, procuramos exemplificar o uso da versão 4 do TASSEL, que é a mais recente e recomendada para GBS (apesar de já existir o TASSEL5). No entanto, em determinado momento, utilizaremos o TASSEL3 porque uma das funções falha na versão que estamos utilizando, de acordo com o fórum do programa na internet. Inclusive você pode – e deve – consultá-lo também sempre que alguma coisa der errado. É bem provável que alguém já tenha passado pelo mesmo problema que você está enfrentando…
Via terminal, baixamos os programas de seus respectivos repositórios, e,
quando foi o caso, descompactamos arquivo com extensão .zip,
renomeando-o convenientemente, como segue:
wget http://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.2.3/bowtie2-2.2.3-linux-x86_64.zip
unzip bowtie2-2.2.3-linux-x86_64.zip
mv bowtie2-2.2.3 bowtie2
git clone git://git.code.sf.net/p/tassel/tassel3-standalone tassel3
git clone git://git.code.sf.net/p/tassel/tassel4-standalone tassel4
Pipeline TASSEL-GBS
A seguir, oferecemos uma sequência de comandos que vai resolver o problema específico deste exemplo, cujos argumentos, na verdade, basearam-se na publicação existente. No entanto, isto não significa que este é o único caminho, tampouco o mais correto e que deva ser seguido em todos os casos. Tenha bastante atenção, sobretudo, se estiver trabalhando com espécies com sistema de cruzamento diferente, ou com populações de trabalhos com outros objetivos. Leia o manual e trabalhos relacionados e consulte o fórum quando começar um novo trabalho!
1º passo: FastqToTagCount
A primeira etapa do processo consiste em contar o número de tags
existentes no arquivo fastq. Isto é realizado utilizando o plugin
FastqToTagCountPlugin.
Argumentos:
-i: diretóriofastqcontendo arquivo(s) bruto(s) de leituras com final_fastq.txtou_fastq.gz;-k: key file;-e: enzima de restrição utilizada para criar a biblioteca;-s: número máximo de leituras boas por lane. Padrão: 300.000.000;-c: número mínimo de vezes que uma tag deve estar presente. Padrão: 1;-o: diretório que conterá o output.cnt(contagem de tags).
Comando:
./tassel4/run_pipeline.pl -Xmx10g -fork1 -FastqToTagCountPlugin -i ./fastq -k D0DTNACXX_1-key-file.tsv -e ApeKI -c 5 -o tagCounts -endPlugin -runfork1 > 1_FastqToTagCount.txt
Perceba que esta etapa é relativamente exigente do ponto de vista computacional (requer ~10 GB de RAM para este conjunto de dados). Então, nós já rodamos esse comando previamente e disponibilizamos o resultado publicamente. Para acessá-lo e movê-lo para a pasta apropriada, bastou:
wget https://dl.dropboxusercontent.com/u/24302457/D0DTNACXX_1.cnt
mv "D0DTNACXX_1.cnt" ./tagCounts/
Além disso, se você quiser salvar o output do terminal em um arquivo,
utilize > para direcionar a saída textual para um .txt, por exemplo.
Esse recurso será omitido nos demais passos porque são mais rápidos, de
tal modo que conseguimos acompanhá-los. Na prática, quando usamos
scripts com todos os comandos e deixamos rodando ad finem, é
recomendável formtemente utilizar este recurso como um log.
2º passo: MergeMultipleTagCountPlugin
A partir daqui, os computadores disponíveis darão conta do recado (pelo
menos, é o que esperamos!). Neste passo, as tags de múltiplos arquivos
em tagCount são combinadas em uma única lista de “master” tags. O
resultado é um arquivo binário (.cnt) que será utilizado pelo plugin
fastqToTBTPlugin para construir os aquivos tags by taxa (TBT).
Argumentos:
-i: diretório que contém os múltiplos arquivos de contagem de tags (.cnt);-o: diretório que conterá o único arquivo de contagem de tags (.cnt);-c: número mínimo de vezes que uma tag deve estar presente. Padrão: 1;-t: especifica uma saída adicional no formato fastq para alinhamento contra genoma de referência.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -MergeMultipleTagCountPlugin -i tagCounts -o mergedTagCounts/myMasterGBSTags.cnt -endPlugin -runfork1
3º passo: TagCountToFastqPlugin
Um passo adicional é necessário na ausência do argumento -t no
plugin anterior. Para ficar mais claro, esse passo foi omitido antes e
essa funcionalidade (conversão de uma lista “master” de contagem de
tags para o formato fastq) é desenvolvida agora pelo plugin
TagCountToFastqPlugin.
Argumentos:
-i: arquivo contendo as contagens de tags (.cnt);-o: arquivo que conterá as tags no formato fastq (.fq);-c: número mínimo de vezes que uma tag deve estar presente. Padrão: 1.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -TagCountToFastqPlugin -i mergedTagCounts/myMasterGBSTags.cnt -o mergedTagCounts/myMasterGBSTags.fq -endPlugin -runfork1
4º passo: bowtie2-build
Essa etapa cria uma série de arquivos necessária na operação do bowtie2.
Argumentos:
- Input: arquivo fasta contendo sequências de bases do genoma (ou de alguma pseudo-referência);
- Output: arquivos com o nome base fornecido.
Comando:
./bowtie2/bowtie2-build ./reference/Osativa_204.fa ./reference/MyReferenceGenome.fa
5º passo: bowtie2
Alinha o conjunto “master” de tags de GBS ao genoma de referência.
Argumentos:
-R X: retorna o melhor alinhamento, se mais de “X + 1” locais são encontrados;-p: número de processadores a ser usado (verifique quantos o seu computador tem usando no “Monitor do sistema” do seu linux). Quanto mais processadores, mais rápido será o alinhamento;--very-sensitive-local: configura a sensibilidade do alinhador;-x: o nome base do genoma de referência;-U: arquivo “master” de contagem de tags no formato fastq (.fq);-S: arquivo que conterá o alinhamento no formato SAM (Sequence Alignment/Map).
Comando:
./bowtie2/bowtie2 -R 4 -p 4 --very-sensitive-local -x ./reference/MyReferenceGenome.fa -U ./mergedTagCounts/myMasterGBSTags.fq -S alignment/myAlignedMasterTags.sam
Ao final, o comando retorna um resumo sobre o alinhamento. Anote os valores obtidos e compare com os dos colegas:
_______ reads; of these:
_______ (___.__%) were unpaired; of these:
_______ (__.__%) aligned 0 times
_______ (__.__%) aligned exactly 1 time
_______ (__.__%) aligned >1 times
__.__% overall alignment rate
6º passo: SAMConverterPlugin
Este plugin converte arquivo de alinhamento no formato SAM (.sam)
produzido pelo bowtie2 em um arquivo binário “tagsOnPhysicalMap”
(.topm).
Argumentos:
-i: arquivo contendo alinhamento (.sam);-o: arquivo que conterá o binário “tagsOnPhysicalMap” (.topm).
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -SAMConverterPlugin -i alignment/myAlignedMasterTags.sam -o topm/myMasterTags.topm -endPlugin -runfork1
7º passo: FastqToTBTPlugin
Este plugin gera um arquivo “TagsByTaxa” para cada arquivo fastq no
diretório que contém as leituras brutas. Para isto, é necessária a lista
“master” de tags no formato .topm ou .cnt.
Argumentos:
-i: diretório contendo os arquivos fastq de leituras brutas de GBS;-k: key file;-e: enzima de restrição utilizada para criar a biblioteca;-o: diretório que conterá o output “TagByTaxa” (.tbt) de contagem de tags por táxon;-c: número mínimo de taxa dentro do arquivo fastq para uma tag ser considerada. Padrão: 1;-t: arquivo “master” contendo contagem das tags (.cnt) de interesse;-m: arquivo contendo “tagsOnPhysicalMap” (.topm). Mutuamente exclusiva com a opção-t;-y: arquivo em formato TBTByte (contagens de 0-127) ao invés de TBTBit (0 ou 1).
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -FastqToTBTPlugin -i ./fastq -k D0DTNACXX_1-key-file.tsv -e ApeKI -o tbt -y -m topm/myMasterTags.topm -endPlugin -runfork1
8º passo: MergeTagsByTaxaFilesPlugin
Combina todos os arquivos .tbt.bin e .tbt.byte presentes nos
diretórios de entrada.
Argumentos:
-i: diretório contendo os arquivos “tagByTaxa” (.tbt.byte);-o: arquivo de saída com extensão.tbt.byte;-s: número máximo de tags no arquivo TBTByte que é considerada durante o processo. Padrão: 200.000.000. Este valor deve ser reduzido em caso de memória RAM limitada;-x: combina contagem de tags para taxa com mesmo nome. Não realizada por padrão.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -MergeTagsByTaxaFilesPlugin -s 50000000 -i tbt -o mergedTBT/myStudy.tbt.byte -endPlugin -runfork1
Agora, os próximos passos consistem da descoberta de SNPs em si e possibilita o output em dois formatos: HAPMAP e VCF. Ambos trazem, basicamente, os mesmos resultados (embora diferenças nas chamadas dos genótipos possam ser verificadas). Porém, enquanto o arquivo HAPMAP traz apenas a informação do SNP para cada indivíduo, o arquivo VCF retorna, adicionalmente, as informações sobre a contagem das leituras que contribuíram para a chamada de genótipos de cada loco. Isto pode ser útil se você mesmo quiser estabelecer seus próprios critérios para considerar se um indivíduo é homozigoto ou heterozigoto, o que, obviamente, vai resultar em algum trabalho adicional.
9º passo: DiscoverySNPCallerPlugin
Alinha as tags da mesma posição física entre si, chama os SNPs de cada alinhamento e retorna os genótipos em um arquivo para cada cromossomo nos formatos VCF ou HAPMAP. Aqui, trabalharemos com o último formato.
Argumentos:
-i: arquivo de entrada do tipo “TagsByTaxa” (TBT) (.tbt.byte);-y: indica que o arquivo de entrada é do tipo TBTByte;-m: arquivo contendo “TagsOnPhysicalMap” (TOPM), ou seja, as posições genômicas das tags;-mUpd: atualiza o arquivo TOPM durante a chamada dos SNPs;-o: arquivo de saída do tipo HAPMAP.+é usado como caractere curinga para substituir o número do cromossomo;-mxSites: número máximo de SNPs por cromossomo. Padrão: 200.000;-mnF: valor mínimo do coeficiente de endogamia ($F = 1 - \frac{H_o}{H_e}$)-p: arquivo opcional de pedigree;-mnMAF: frequência mínimo do alelo de menor frequência. Padrão: 0,01;-mnMAC: valor mínimo de contagem do alelo de menor frequência. Padrão: 10;-mnLCov: frequência mínimo de cobertura (proporção de taxa com pelo menos uma tag para o SNP). Padrão: 0,1;-errRate: frequência médio da taxa de erro de sequenciamento. Padrão: 0,01;-ref: caminho para o genoma de referência, caso queira usá-lo para definir o alelo de referência;-inclRare: inclusão de alelos raros (3º e 4º estados alélicos);-inclGaps: inclusão de gaps (InDels);-callBiSNPsWGap: inclusão de gap como 3º estado, quando existir;-sC: primeiro cromossomo.-eC: último cromossomo.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -DiscoverySNPCallerPlugin -i mergedTBT/myStudy.tbt.byte -y -m topm/myMasterTags.topm -mUpd topm/myMasterTagsWithVariants.topm -o hapmap/raw/myGBSGenos_chr+.hmp.txt -mnF 0.9 -mnMAF 0.005 -mnMAC 20 -ref ./reference/MyReferenceGenome.fa -sC 1 -eC 12 -endPlugin -runfork1
./tassel4/run_pipeline.pl -Xmx30g -fork1 -DiscoverySNPCallerPlugin -i mergedTBT/myStudy.tbt.byte -y -m topm/myMasterTags.topm -mUpd topm/myMasterTagsWithVariants.topm -o hapmap/raw/myGBSGenos_chr+.hmp.txt -vcf -mnMAF 0.01 -mnMAC 999 -sC 1 -eC 5616 -endPlugin -runfork1 > 9_DiscoverySNPCallerDiscovery.txt
10º passo: MergeDuplicateSNPsPlugin
Combina SNPs duplicados de acordo com um limite de mismatches.
Argumentos:
-hmp: arquivo HAPMAP de entrada (.hmp.txt).+é usado como caractere curinga para substituir o número do cromossomo;-o: arquivo HAPMAP de saída;-misMat: limite de mismatches a partir do qual SNPs duplicados não serão combinados. Padrão: 0,05;-p: arquivo opcional de pedigree;-callHets: chamada de genótipo como heterozigoto quando dois SNPs duplicados discordam entre si;-kpUnmergDups: mantém SNPs duplicados quando não são combinados. Padrão: deletá-los;-sC: primeiro cromossomo;-eC: último cromossomo.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -MergeDuplicateSNPsPlugin -hmp hapmap/raw/myGBSGenos_chr+.hmp.txt -o hapmap/mergedSNPs/myGBSGenos_mergedSNPs_chr+.hmp.txt -sC 1 -eC 12 -endPlugin -runfork1
Ao final, o número de SNPs remanescentes é indicado. Anote-o:
Number of sites written after deleting any remaining, unmerged duplicate SNPs: _____
11º passo: MergeIdenticalTaxaPlugin
Combina as diferentes chamadas de genótipos para as amostras com o mesmo
nome (até o 1º :).
Argumentos:
-hmp: arquivo HAPMAP de entrada (.hmp.txt).+é usado como caractere curinga para substituir o número do cromossomo;-o: arquivo HAPMAP de saída;-xHets: exclusão das chamadas de heterozigotos;-hetFreq: frequência de heterozigotos. Padrão: 0,8;-sC: primeiro cromossomo;-eC: último cromossomo.
Comando:
./tassel3/run_pipeline.pl -Xmx2g -fork1 -MergeIdenticalTaxaPlugin -hmp hapmap/mergedSNPs/myGBSGenos_mergedSNPs_chr+.hmp.txt -o hapmap/mergedTaxa/myGBSGenos_mergedSNPs_mergedTaxa_chr+.hmp.txt -xHets -sC 1 -eC 12 -endPlugin -runfork1
Ao final, o comando retorna um resumo sobre o número de taxa combinados. Anote os valores obtidos e compare com os dos colegas:
Total taxa: ___
Unique taxa: ___
12º passo: GBSHapMapFiltersPlugin
Há um passo adicional de filtragem que você pode fazer utilizando o
TASSEL, mas que serve apenas para o formato HAPMAP. Para trabalhar
com o formato VCF, entre diretamente com o arquivo no TASSEL com
interface (para acioná-lo, via terminal, digite:
./tassel4/start_tassel.pl -Xmx2g) ou, alternativamente, utilize o
VCFTools ou seus próprios
comandos em R (como será demonstrado posteriormente).
Para nosso exemplo, utilizaremos um MAF de 0,25 já que se trata de uma população de mapeamento. Assim, já descartamos os locos com elevada distorção da segregação esperada (que para RILs é de 1:1).
Argumentos:
-hmp: arquivo HAPMAP de entrada (.hmp.txt).+é usado como caractere curinga para substituir o número do cromossomo;-o: arquivo HAPMAP de saída;-mnTCov: valor mínimo de cobertura de taxa (proporção de amostras com chamadas diferente deNpara o SNP). Padrão: sem filtro;-mnSCov: valor mínimo de cobertura de SNPs (proporção de SNPs chamados para determinada amostra). Padrão: sem filtro;-mnF: valor mínimo do coeficiente de endogamia F;-p: arquivo opcional de pedigree;-mnMAF: frequência mínima do alelo de menor frequência. Padrão: 0,01;-mxMAF: frequência máxima do alelo de menor frequência. Padrão: 1 (sem filtro);-hLD: filtra SNPs com significância estatística para desequilíbrio de ligação (LD) com um SNP vizinho. Padrão: sem filtro;-mnR2: valor mínimo de R2 para o filtro de LD. Padrão: 0,01;-mnBonP: valor mínimo de p-valor corrigido utilizando o critério de Bonferroni. Padrão: 0,01;-sC: primeiro cromossomo;-eC: último cromossomo.
Comando:
./tassel4/run_pipeline.pl -Xmx2g -fork1 -GBSHapMapFiltersPlugin -hmp hapmap/mergedTaxa/myGBSGenos_mergedSNPs_mergedTaxa_chr+.hmp.txt -o hapmap/filt/myGBSGenos_mergedSNPsFilt_chr+.hmp.txt -mnTCov 0.1 -mnSCov 0.8 -mnMAF 0.25 -mxMAF 1 -sC 1 -eC 12 -endPlugin -runfork1
Extra: R/qtl
Utilizaremos o arquivo HAPMAP filtrado para, rapidamente, demonstrar o uso das saídas de cada cromossomo para estimar um mapa genético para a população genotipada. Esta etapa exigirá um pouco de conhecimento do R e consistirá apenas de uma exibição. Obviamente, nada o impede de tentar rodar os comandos posteriormente. Para uma breve exibição da qualidade dos marcadores já ordenados com base no genoma, utilizaremos um pacote do R denominado R/qtl.
Conversão dos arquivos HAPMAP para formato MAPMAKER/EXP
Para populações experimentais baseadas em cruzamento de linhagens endogâmicas (como RILs, retrocruzamentos ou F2), o R/qtl aceita o formato do MAPMAKER/EXP, mas para o caso de RILs, o arquivo deve ser entrado tal qual populações de retrocruzamentos e, depois, convertidos com uma função interna.
Inicialmente, lemos os dados de SNPs oriundos de cada cromossomo e os
combinamos em um único objeto chamado dados. Mas antes, configure um
caminho para o seu diretório que contém os arquivos. Assim:
setwd("/home/rramadeu/Documentos/tassel-tutorial/gbs-tassel-arroz/hapmap/filt")
chr1 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr1.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr2 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr2.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr3 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr3.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr4 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr4.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr5 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr5.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr6 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr6.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr7 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr7.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr8 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr8.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr9 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr9.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr10 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr10.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr11 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr11.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
chr12 <- read.table(file="myGBSGenos_mergedSNPsFilt_chr12.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
rbind(dim(chr1)[1], dim(chr2)[1], dim(chr3)[1], dim(chr4)[1], dim(chr5)[1], dim(chr6)[1], dim(chr7)[1], dim(chr8)[1], dim(chr9)[1], dim(chr10)[1], dim(chr11)[1], dim(chr12)[1])
## [,1]
## [1,] 1200
## [2,] 837
## [3,] 777
## [4,] 809
## [5,] 595
## [6,] 693
## [7,] 631
## [8,] 581
## [9,] 539
## [10,] 513
## [11,] 697
## [12,] 468
dados <- rbind(chr1, chr2, chr3, chr4, chr5, chr6, chr7, chr8, chr9, chr10, chr11, chr12)
dim(dados)
## [1] 8340 184
Alguma formatação para o cabeçalho do arquivo faz-se necessária por questões de organização. Veja:
colnames(dados)[1:22]
## [1] "alleles" "chrom"
## [3] "pos" "strand"
## [5] "assembly." "center"
## [7] "protLSID" "assayLSID"
## [9] "panelLSID" "QCcode"
## [11] "Azucena.MERGE" "IR64.MERGE"
## [13] "IR64xAzu_100.D0DTNACXX.1.1.G07" "IR64xAzu_102.D0DTNACXX.1.1.H07"
## [15] "IR64xAzu_106.D0DTNACXX.1.1.A08" "IR64xAzu_107.D0DTNACXX.1.1.B08"
## [17] "IR64xAzu_109.D0DTNACXX.1.1.C08" "IR64xAzu_111.D0DTNACXX.1.1.D08"
## [19] "IR64xAzu_112.D0DTNACXX.1.1.E08" "IR64xAzu_115.D0DTNACXX.1.1.F08"
## [21] "IR64xAzu_116.D0DTNACXX.1.1.G09" "IR64xAzu_120.D0DTNACXX.1.1.H09"
dados <- dados[c(11:183)]
colnames(dados)[1:2] <- matrix(unlist(strsplit(colnames(dados)[c(1:2)], "\\.")), ncol=2, byrow=T)[,1]
colnames(dados)[3:173] <- matrix(unlist(strsplit(colnames(dados)[c(3:173)], "\\.")), ncol=5, byrow=T)[,1]
colnames(dados)[1:12]
## [1] "Azucena" "IR64" "IR64xAzu_100" "IR64xAzu_102"
## [5] "IR64xAzu_106" "IR64xAzu_107" "IR64xAzu_109" "IR64xAzu_111"
## [9] "IR64xAzu_112" "IR64xAzu_115" "IR64xAzu_116" "IR64xAzu_120"
Os passos a seguir basicamente darão conta de converter as chamadas de
SNPs para cada indivíduo no formato HAPMAP para o formato requerido para
mapeamento. Nesse caso, a população de RILs deve apresentar, ao invés da
representação de bases nitrogenadas, as letras A para designar,
digamos, o alelo que veio do genitor “Azucena”, e H para designar o
alelo que veio do genitor “IR64” (para o caso do arquivo para o
R/qtl). Segue:
dados[c(1:6),c(1:11)]
## Azucena IR64 IR64xAzu_100 IR64xAzu_102 IR64xAzu_106 IR64xAzu_107
## S1_190590 C T C N T C
## S1_205765 A G A N G A
## S1_212589 G C G G C G
## S1_242949 A C N A C A
## S1_252897 A G A N G A
## S1_289231 T C T T C N
## IR64xAzu_109 IR64xAzu_111 IR64xAzu_112 IR64xAzu_115 IR64xAzu_116
## S1_190590 N T T T C
## S1_205765 N N G G A
## S1_212589 N C C C G
## S1_242949 A C C C A
## S1_252897 A G G G A
## S1_289231 T C C C T
dados <- subset(dados, Azucena == "A" | Azucena == "T" | Azucena == "C" | Azucena == "G")
dados <- subset(dados, IR64 == "A" | IR64 == "T" | IR64 == "C" | IR64 == "G")
dim(dados)
## [1] 8221 173
lista <- lapply(split(dados[3:ncol(dados)], f=1:nrow(dados)), matrix, ncol=171, nrow=1)
for(i in 1:nrow(dados)) {
for(j in 1:171) {
if(lista[[i]][j] == dados[i,"Azucena"]) {
lista[[i]][j] <- "A"
} else if(lista[[i]][j] == dados[i,"IR64"]) {
lista[[i]][j] <- "H"
} else {
lista[[i]][j] <- "-"
}
}
}
dados.mm <- as.data.frame(matrix(unlist(lista), ncol = 171, byrow = TRUE))
rownames(dados.mm) <- rownames(dados)
colnames(dados.mm) <- colnames(dados)[3:173]
dados.mm[c(1:6),c(1:10)]
## IR64xAzu_100 IR64xAzu_102 IR64xAzu_106 IR64xAzu_107 IR64xAzu_109
## S1_190590 A - H A -
## S1_205765 A - H A -
## S1_212589 A A H A -
## S1_242949 - A H A A
## S1_252897 A - H A A
## S1_289231 A A H - A
## IR64xAzu_111 IR64xAzu_112 IR64xAzu_115 IR64xAzu_116 IR64xAzu_120
## S1_190590 H H H A A
## S1_205765 - H H A A
## S1_212589 H H H A A
## S1_242949 H H H A A
## S1_252897 H H H A A
## S1_289231 H H H A A
Como a persistência de heterozigotos pode ter alterado a frequência de dados perdidos, investigamos a proporção destes e eliminamos os locos com mais de 80% de dados perdidos. Do mesmo modo, excluímos eventuais locos duplicados que, para a análise de ligação, não acrescenta informação e, ao invés, aumenta o tempo de processamento. Assim:
excluir <- c()
for(i in 1:nrow(dados.mm)) {
if(table(t(dados.mm[i,]))["-"] > (ncol(dados.mm)*0.2)) {
excluir <- c(excluir, i)
}
}
length(excluir)
## [1] 847
dados.mm <- dados.mm[-excluir,]
dados.mm <- dados.mm[!duplicated(dados.mm),]
dim(dados.mm)
## [1] 5981 171
Finalmente, construindo o arquivo .raw no formato MAPMAKER/EXP e
um .txt com o pré-agrupamento dos marcadores, vem:
number.markers <- dim(dados.mm)[1]
number.individ <- dim(dados.mm)[2]
marker.genot <- as.matrix(dados.mm)
marker.names <- matrix(rownames(dados.mm), nrow=number.markers, ncol=1)
outfile <- "hapmap"
write.table(c("data type f2 backcross"), paste(outfile,".raw", sep=""), append=F, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
write.table(t(c(number.individ, number.markers, 0)), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
for(i in 1:number.markers) {
write.table(t(c(paste("*", marker.names[i], sep=""), marker.genot[i,], sep="")), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
}
for(i in 1:number.markers) {
write.table(paste(strsplit(strsplit(marker.names[i], "_")[[1]][1], "S")[[1]][2], marker.names[i], sep=" "), "hapmap.map", append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
}
R/qtl
No R/qtl, fazemos a leitura dos dados e do arquivo contendo os marcadores ordenados construídos anteriormente, convertemos o arquivo então no formato de retrocruzamentos para o formato de RILs e, finalmente, estimamos o mapa. Assim:
library(qtl)
hmp.cross <- read.cross(format ="mm", dir="/home/rramadeu/Documentos/tassel-tutorial/gbs-tassel-arroz/hapmap/filt", file="hapmap.raw", mapfile="hapmap.map", estimate.map=F)
## --Read the following data:
## Type of cross: bc
## Number of individuals: 171
## Number of markers: 5980
## Number of phenotypes: 0
## Warning in summary.cross(cross): Some chromosomes > 1000 cM in length; there may be a problem with the genetic map.
## (Perhaps it is in basepairs?)
## --Cross type: bc
hmp.cross <- convert2riself(hmp.cross)
hmp.map <- est.map(hmp.cross, map.function="kosambi", n.cluster=8, tol=0.01, maxit=1000)
## -Running est.map via a cluster of 8 nodes.
O gráfico a seguir mostra as proporções de alelos segregando 1:1 nas RILs (vermelho e azul). Pontos brancos são dados perdidos.
geno.image(hmp.cross)
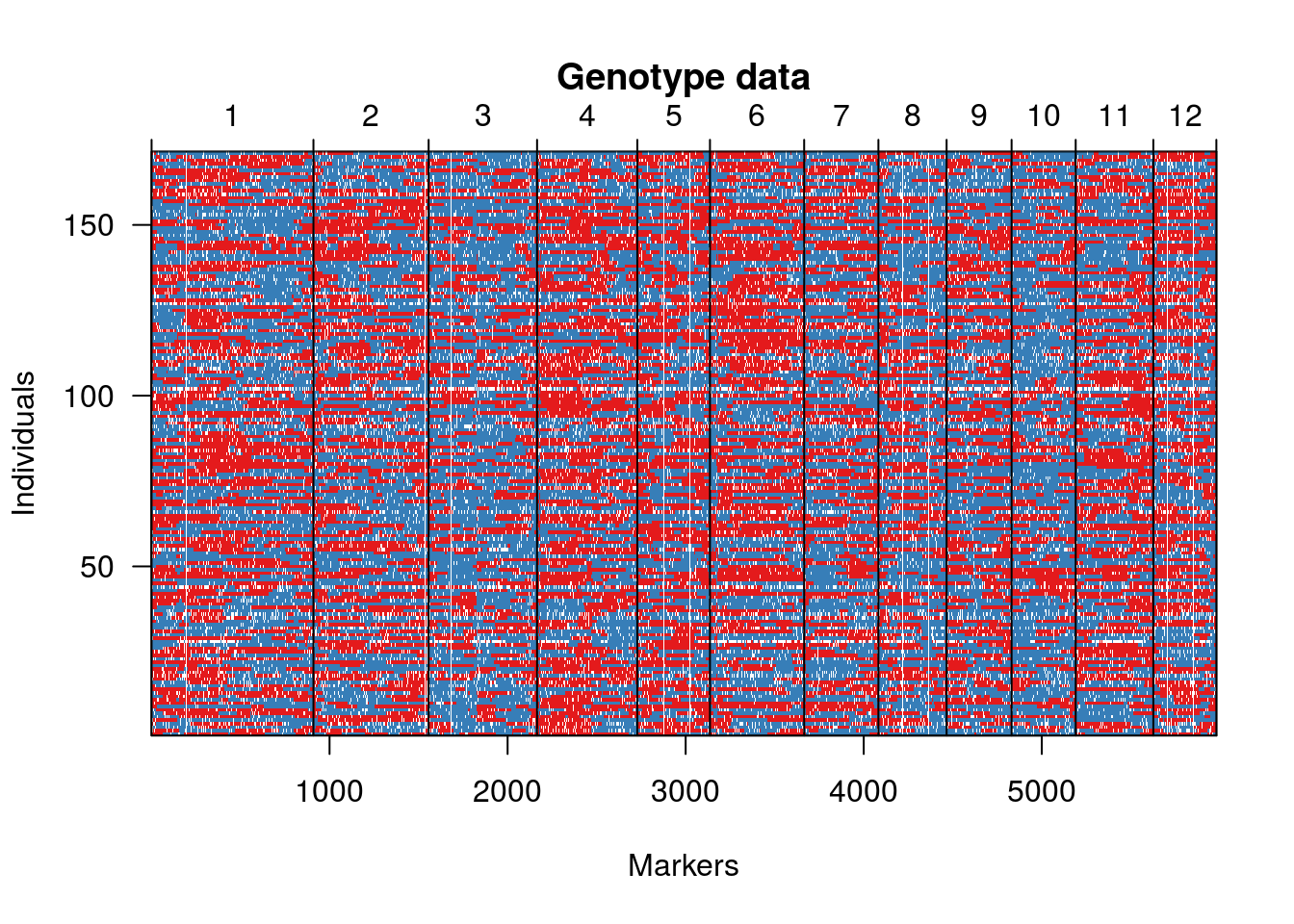
Agora, segue uma representação gráfica dos marcadores em ordem pré-estabelecida. Neste caso, utilizamos a ordenação existente com base no genoma de referência. Usualmente, essa ordem deve ser estimada.
hmp.cross <- replace.map(hmp.cross, hmp.map)
plot.map(hmp.cross, show.marker.names=FALSE)
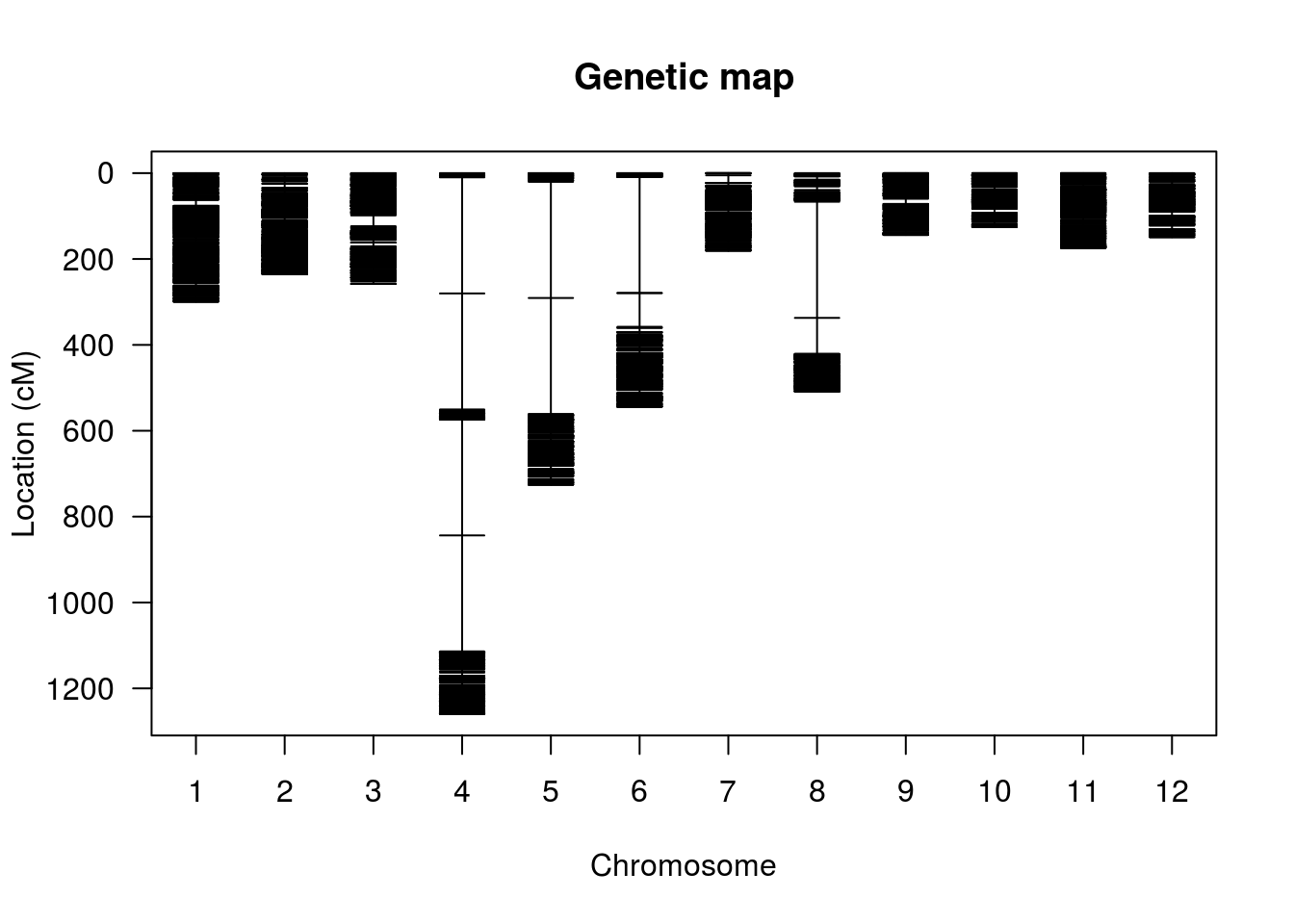
Finalmente, uma representação útil da relação de desequilíbrio de ligação entre os marcadores é fornecido por um heatmap:
plotRF(hmp.cross)
## Warning in plotRF(hmp.cross): Running est.rf.
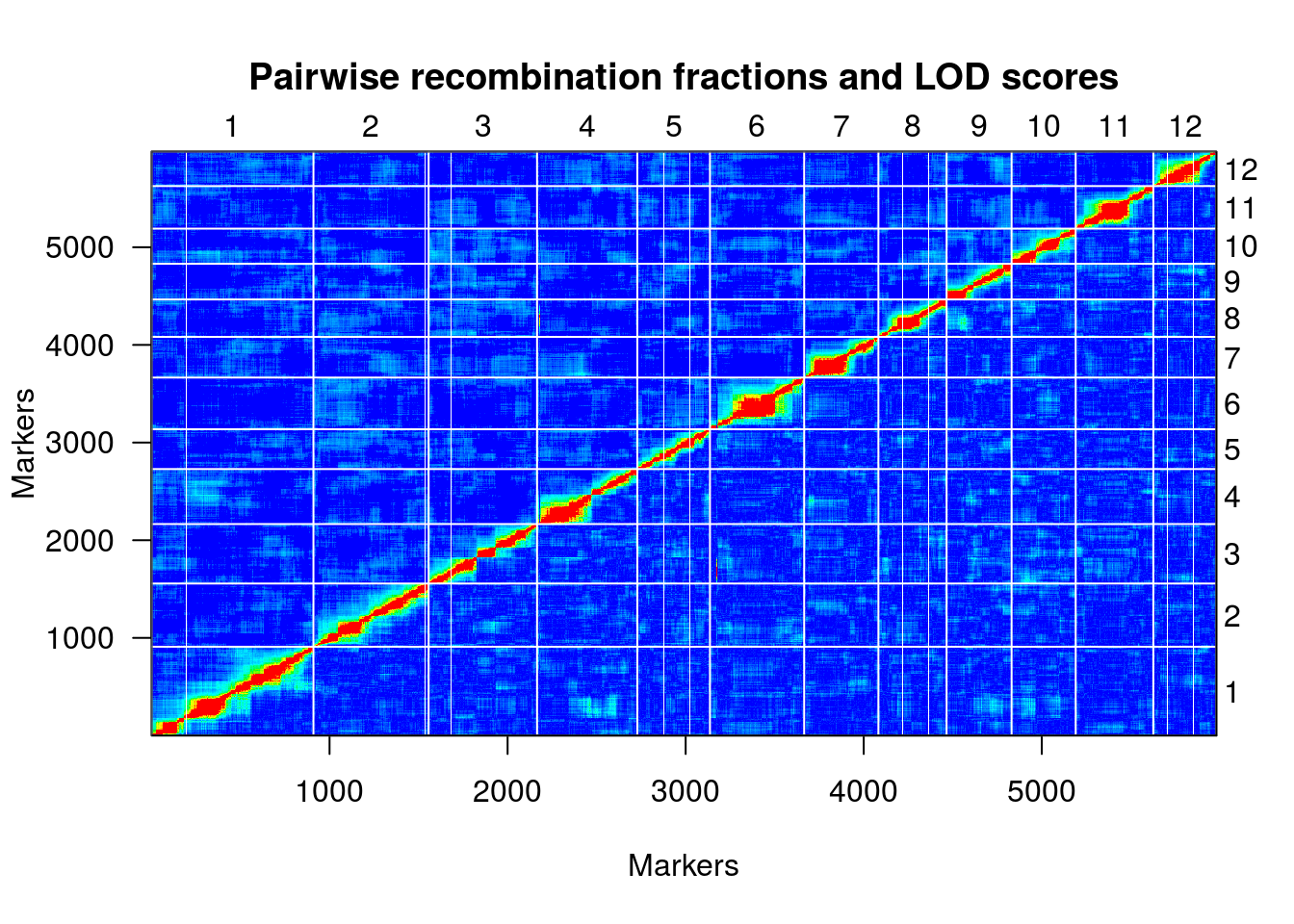
table(formLinkageGroups(hmp.cross, max.rf=0.35, min.lod=8)[,2])
Warning in formLinkageGroups(hmp.cross, max.rf = 0.35, min.lod = 8):
Running est.rf.
##
## 1 2 3 4 5 6 7 8 9 10 11 12
## 978 909 648 527 436 407 383 365 362 354 341 270
hmp.map <- formLinkageGroups(hmp.cross, max.rf=0.35, min.lod=8, reorgMarkers=TRUE)
## Warning in formLinkageGroups(hmp.cross, max.rf = 0.35, min.lod = 8,
## reorgMarkers = TRUE): Running est.rf.
plotRF(hmp.map)
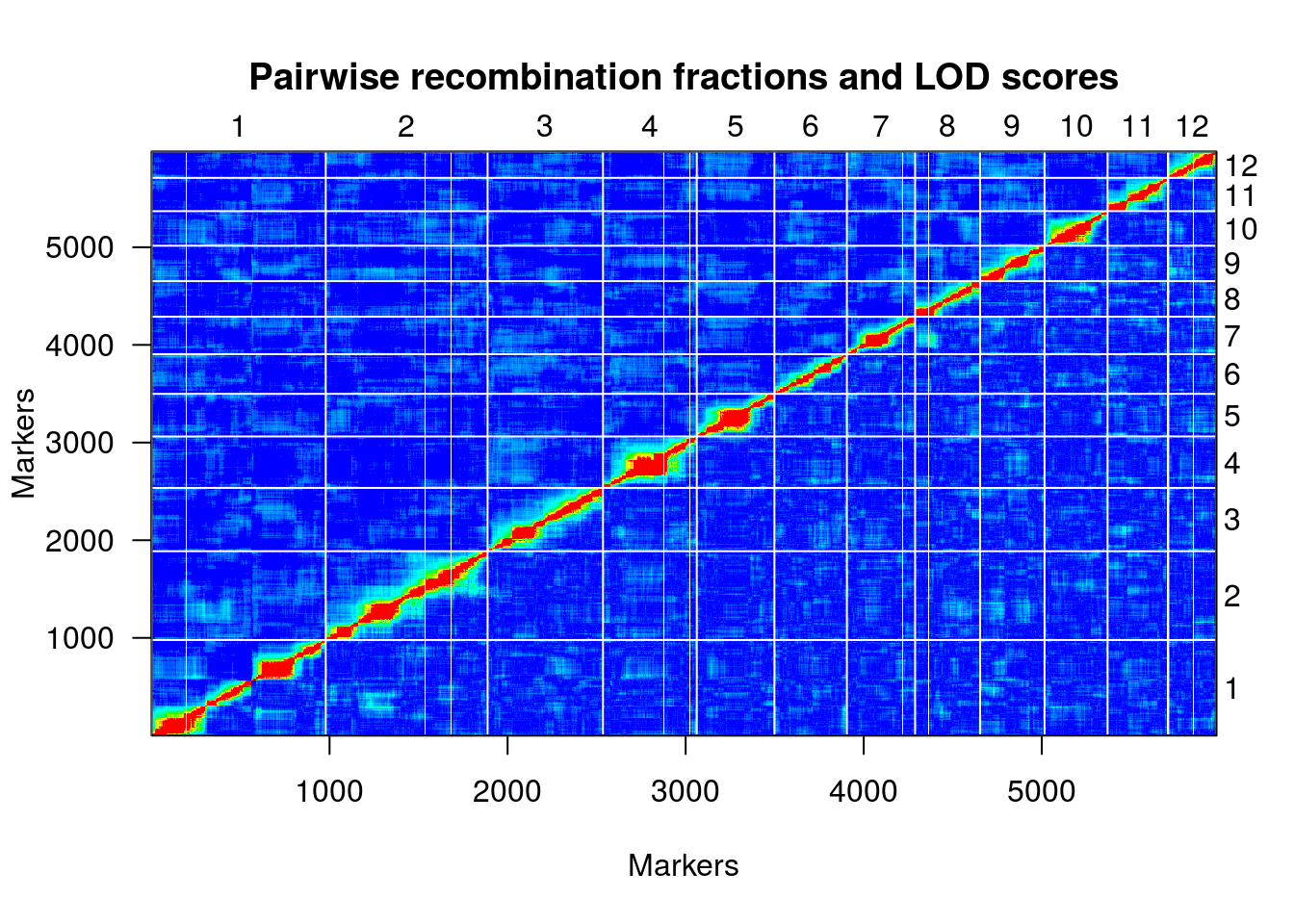
hmp.map <- orderMarkers(hmp.map, chr=c(1:12), use.ripple=F, map.function="kosambi", maxit=1000, tol=0.01, sex.sp=F)
plotRF(hmp.map)
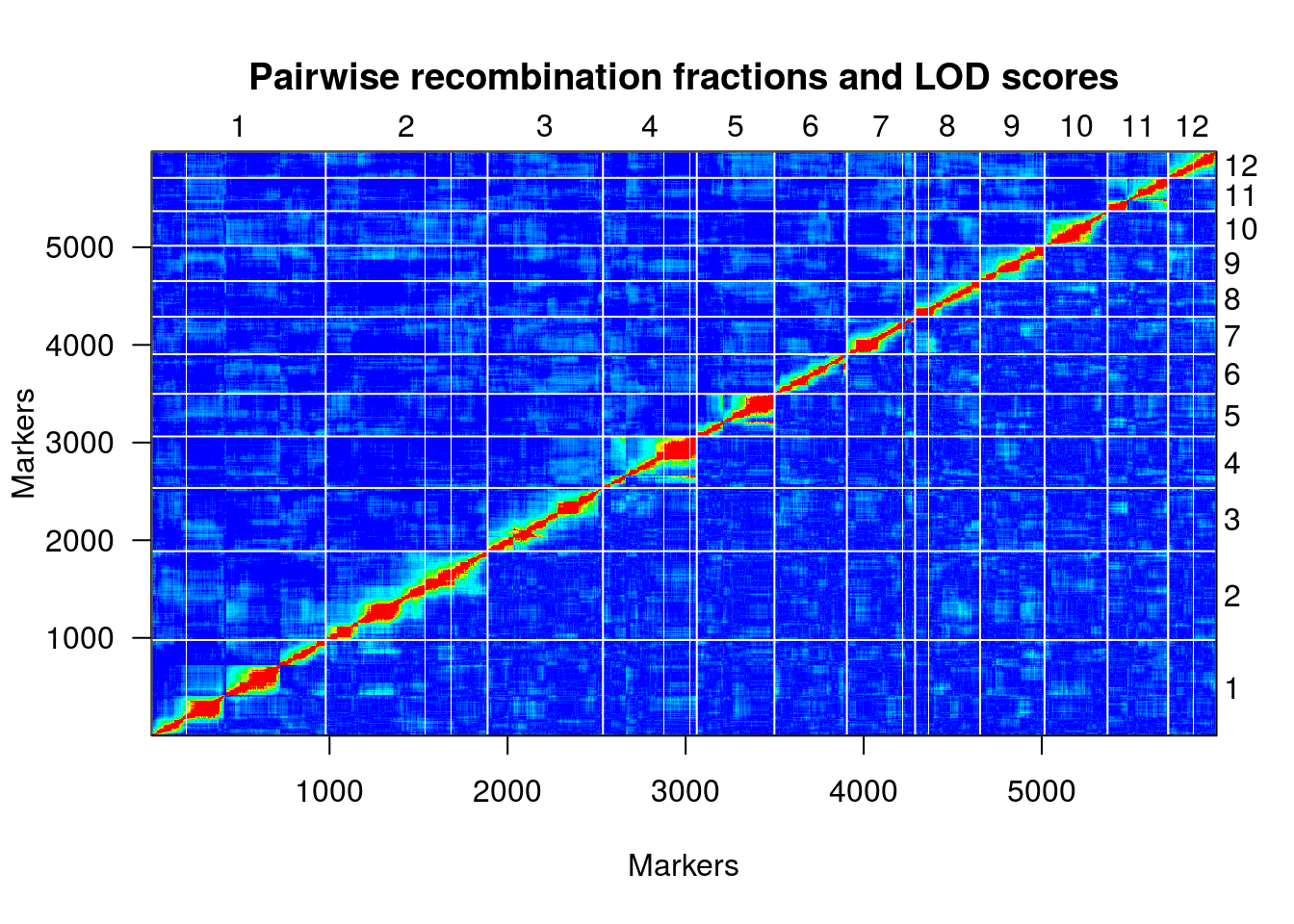
plotMap(hmp.map, show.marker.names=FALSE)
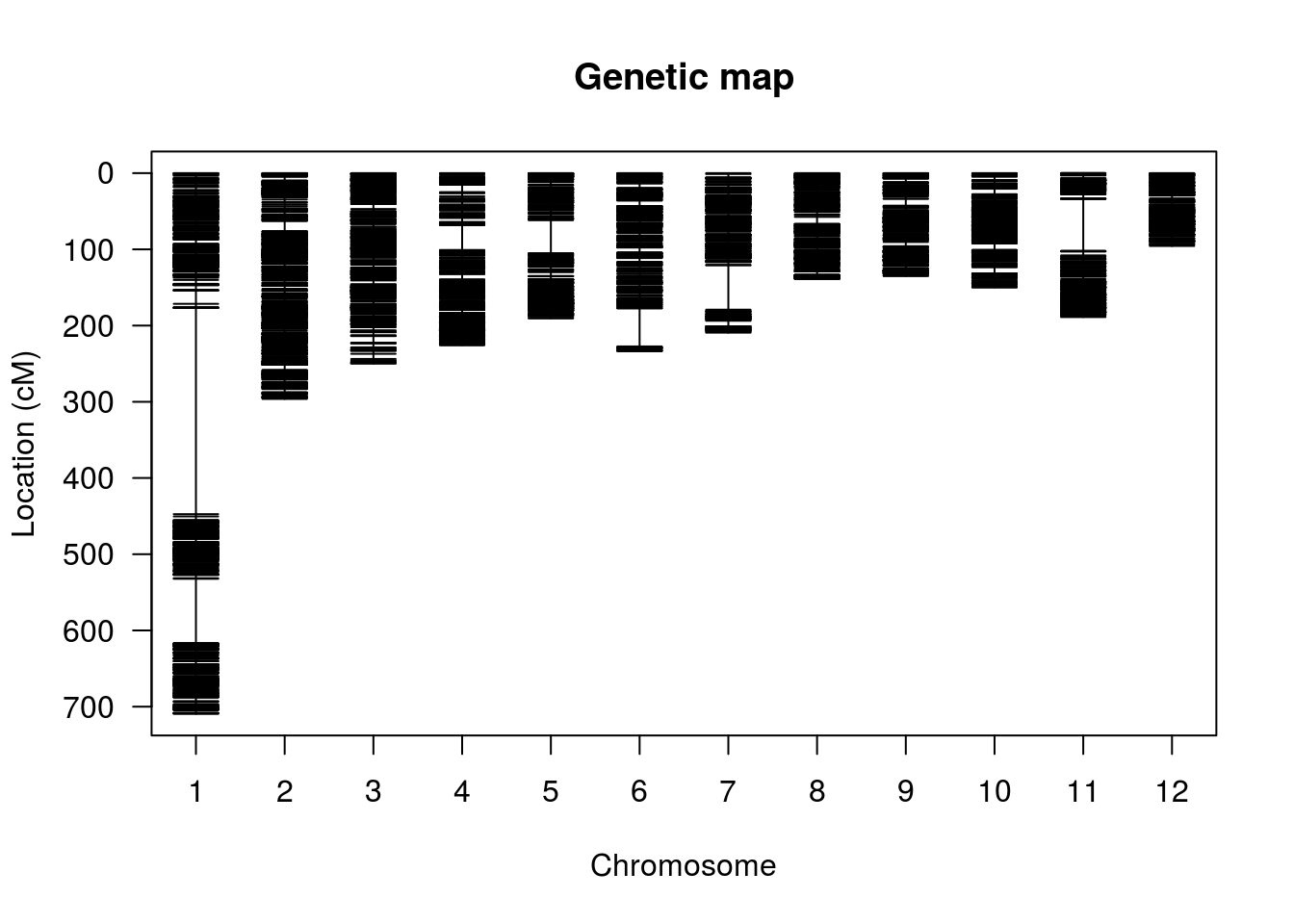
summaryMap(hmp.map)
## n.mar length ave.spacing max.spacing
## 1 978 709.4 0.7 270.5
## 2 909 296.3 0.3 13.3
## 3 648 249.9 0.4 9.0
## 4 527 225.6 0.4 32.7
## 5 436 190.5 0.4 43.7
## 6 407 233.4 0.6 50.1
## 7 383 209.3 0.5 58.2
## 8 365 138.8 0.4 9.3
## 9 362 135.0 0.4 9.1
## 10 354 149.8 0.4 8.5
## 11 341 188.9 0.6 68.4
## 12 270 95.5 0.4 3.7
## overall 5980 2822.2 0.5 270.5
Pipeline UNEAK
O pipeline Universal Network Enabled Analysis Kit (UNEAK) foi criado pelo mesmo grupo do TASSEL e serve para GBS de organismos sem genoma de referência. Ele foi apresentado neste artigo; por favor, refira-se a ele e ao tutorial completo quando utilizar este programa. Como dito anteriormente, a função didática deste material restringe bastante as informações contidas no tutorial; consulte-o sempre, então. As versões que compõem o TASSEL já trazem as funções deste pipeline, de tal modo que tendo executado o exemplo anterior, você já será capaz de executar os passos a seguir.
1º passo: UCreatWorkingDirPlugin
Do mesmo modo que o TASSEL-GBS, este pipeline requer a
participação de vários diretórios. Agora, um plugin específico,
chamado UCreatWorkingDirPlugin, foi designado para realização
automática desta etapa.
Argumento:
-w: diretório de trabalho que conterá os subdiretórios.
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UCreatWorkingDirPlugin -w ./uneak/ -endPlugin -runfork1
Porque o pipeline requer, copiamos o arquivo das pastas /fastq e o
key file para /uneak/Illumina e /uneak/key, criada por este
comando anterior. Assim:
cp ./fastq/* ./uneak/Illumina/
cp D0DTNACXX_1-key-file.tsv ./uneak/key/
Pode ser útil lembrar de deletar alguma das cópias dos arquivos contendo leituras após acabar de rodar o pipeline, porque esses arquivos costumam grande e, por isso, ocupam bastante espaço.
2º passo: UFastqToTagCountPlugin
Este plugin coloca no diretório ./tagCounts listas de contagem de
tags para cada amostra no foramto .cnt.
Argumentos:
-w: diretório de trabalho;-e: enzima utilizada na construção da(s) biblioteca(s) sequenciada(s).
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UFastqToTagCountPlugin -w ./uneak/ -e ApeKI -endPlugin -runfork1
3º passo: UMergeTaxaTagCountPlugin
Agora, contagem de tags presentes em arquivos diferentes serão
reunidos para o mesmo táxon em um arquivo “master tagCount” .cnt a ser
armazenado na pasta ./mergedTagCounts.
Argumentos:
-w: diretório de trabalho;-c: contagem mínima de tags a ser considerada. Padrão: 5.
Comandos:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UMergeTaxaTagCountPlugin -w ./uneak/ -c 5 -endPlugin -runfork1
4º passo: UTagCountToTagPairPlugin
Identifica pares de tags para chamada de SNP via filtro de network.
Argumentos:
-w: diretório de trabalho;-e: tolerância do erro no filtro de network. Padrão: 0,03.
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UTagCountToTagPairPlugin -w ./uneak/ -e 0.03 -endPlugin -runfork1
5º passo: UTagPairToTBTPlugin
Agora, arquivo “TagsByTaxa” é gerado para as tags presentes no arquivo “tagPair”. Como apenas “tagsByTaxaByte” é suportado pelo pipeline, existe um limite de 127 tags.
Argumento:
-w: diretório de trabalho.
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UTagPairToTBTPlugin -w ./uneak/ -endPlugin -runfork1
Observe o total de “TagsByTaxa”. Compare este valor com aquele do pipeline dependente de referência e imagine qual a razão da diferença, se houver.
______ tags will be output to tbt.bin
6º passo: UTBTToMapInfoPlugin
Gera um arquivo “mapInfo” para uma saída do tipo HAPMAP.
Argumento:
-w: diretório de trabalho.
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UTBTToMapInfoPlugin -w ./uneak/ -endPlugin -runfork1
7º passo: UMapInfoToHapMapPlugin
Libera uma saída do tipo HAPMAP propriamente dita
Argumentos:
-w: diretório de trabalho;-mnMAF: mínima menor frequência alélia. Padrão: 0,05;-mxMAF: máxima menor frequência alélica. Padrão: 0,5;-mnC: mínima taxa de chamada (proporção de taxa coberta por pelo menos uma tag);-mxC: máxima taxa de chamada. Padrão: 1.
Comando:
./tassel3/run_pipeline.pl -Xmx10g -fork1 -UMapInfoToHapMapPlugin -w ./uneak/ -mnMAF 0.3 -mxMAF 0.5 -mnC 0 -mxC 1 -endPlugin -runfork1
Anote o total de SNPs resultantes deste pipeline. Houve prejuízo desta quantidade quando comparada ao total de polimorfismos resultantes do pipeline dependente de referência?
HapMap file is written. There are ______ SNPs in total
Extra: R/qtl com dados do UNEAK
setwd("/home/rramadeu/Documentos/tassel-tutorial/gbs-tassel-arroz/uneak/hapMap/")
uneak <- read.table(file="HapMap.hmp.txt", header=TRUE, comment.char="", sep="\t", row.names=1, stringsAsFactors=FALSE)
dim(uneak)
## [1] 57163 184
colnames(uneak)[c(1:12,180:183)]
## [1] "alleles"
## [2] "chrom"
## [3] "pos"
## [4] "strand"
## [5] "assembly"
## [6] "center"
## [7] "protLSID"
## [8] "assayLSID"
## [9] "panelLSID"
## [10] "QCcode"
## [11] "Azucena_merged_X3"
## [12] "IR64_merged_X3"
## [13] "IR64xAzu_307_D0DTNACXX_1_2_G07_X5"
## [14] "IR64xAzu_355_D0DTNACXX_1_2_G09_X5"
## [15] "IR64xAzu_120_D0DTNACXX_1_1_H09_X5"
## [16] "IR64xAzu_74_D0DTNACXX_1_1_G05_X5"
uneak <- uneak[c(11:183)]
dim(uneak)
## [1] 57163 173
colnames(uneak)[1:2] <- matrix(unlist(strsplit(colnames(uneak)[1:2], "_")), ncol=3, byrow=T)[,1]
colnames(uneak)[3:173] <- paste("IR64xAzu_", matrix(unlist(strsplit(colnames(uneak)[3:173], "_")), ncol=7, byrow=T)[,2], sep = "")
colnames(uneak)[c(1:12,170:173)]
## [1] "Azucena" "IR64" "IR64xAzu_330" "IR64xAzu_191"
## [5] "IR64xAzu_81" "IR64xAzu_377" "IR64xAzu_54" "IR64xAzu_80"
## [9] "IR64xAzu_62" "IR64xAzu_207" "IR64xAzu_89" "IR64xAzu_266"
## [13] "IR64xAzu_307" "IR64xAzu_355" "IR64xAzu_120" "IR64xAzu_74"
uneak[c(1:6),c(1:11)]
## Azucena IR64 IR64xAzu_330 IR64xAzu_191 IR64xAzu_81 IR64xAzu_377
## TP3 N N T N N N
## TP4 C A C A N N
## TP6 N N N N N N
## TP7 T A T A N A
## TP8 T C N N N N
## TP9 T G T T T T
## IR64xAzu_54 IR64xAzu_80 IR64xAzu_62 IR64xAzu_207 IR64xAzu_89
## TP3 N N N N N
## TP4 N N N N N
## TP6 N N N N N
## TP7 N N N N N
## TP8 N N N N N
## TP9 T N T G G
uneak <- subset(uneak, Azucena == "A" | Azucena == "T" | Azucena == "C" | Azucena == "G")
uneak <- subset(uneak, IR64 == "A" | IR64 == "T" | IR64 == "C" | IR64 == "G")
dim(uneak)
## [1] 31296 173
lista <- lapply(split(uneak[3:ncol(uneak)], f=1:nrow(uneak)), matrix, ncol=171, nrow=1)
for(i in 1:nrow(uneak)) {
for(j in 1:171) {
if(lista[[i]][j] == uneak[i,"Azucena"]) {
lista[[i]][j] <- "A"
} else if(lista[[i]][j] == uneak[i,"IR64"]) {
lista[[i]][j] <- "H"
} else {
lista[[i]][j] <- "-"
}
}
}
uneak.mm <- as.data.frame(matrix(unlist(lista), ncol = 171, byrow = TRUE))
rownames(uneak.mm) <- rownames(uneak)
colnames(uneak.mm) <- colnames(uneak)[3:173]
uneak.mm[c(1:6),c(1:10)]
## IR64xAzu_330 IR64xAzu_191 IR64xAzu_81 IR64xAzu_377 IR64xAzu_54
## TP4 A H - - -
## TP7 A H - H -
## TP8 - - - - -
## TP9 A A A A A
## TP12 A - - - A
## TP15 A - - - -
## IR64xAzu_80 IR64xAzu_62 IR64xAzu_207 IR64xAzu_89 IR64xAzu_266
## TP4 - - - - A
## TP7 - - - - H
## TP8 - - - - -
## TP9 - A H H A
## TP12 A H - - H
## TP15 A A - A -
excluir <- c()
for(i in 1:nrow(uneak.mm)) {
if(table(t(uneak.mm[i,]))["-"] > (ncol(uneak.mm)*0.2)) {
excluir <- c(excluir, i)
}
}
length(excluir)
## [1] 28502
uneak.mm <- uneak.mm[-excluir,]
uneak.mm <- uneak.mm[!duplicated(uneak.mm),]
dim(uneak.mm)
## [1] 2794 171
number.markers <- dim(uneak.mm)[1]
number.individ <- dim(uneak.mm)[2]
marker.genot <- as.matrix(uneak.mm)
marker.names <- matrix(rownames(uneak.mm), nrow=number.markers, ncol=1)
outfile <- "uneak"
write.table(c("data type f2 backcross"), paste(outfile,".raw", sep=""), append=F, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
write.table(t(c(number.individ, number.markers, 0)), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
for(i in 1:number.markers) {
write.table(t(c(paste("*", marker.names[i], sep=""), marker.genot[i,], sep="")), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
}
for(i in 1:number.markers) {
write.table(paste(1, marker.names[i], sep=" "), "uneak.map", append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
}
library(qtl)
une.cross <- read.cross(format ="mm", dir="/home/rramadeu/Documentos/tassel-tutorial/gbs-tassel-arroz/uneak/hapMap", file="uneak.raw", mapfile="uneak.map", estimate.map=F)
## --Read the following data:
## Type of cross: bc
## Number of individuals: 171
## Number of markers: 2794
## Number of phenotypes: 0
## Warning in summary.cross(cross): Some chromosomes > 1000 cM in length; there may be a problem with the genetic map.
## (Perhaps it is in basepairs?)
## --Cross type: bc
une.cross <- convert2riself(une.cross)
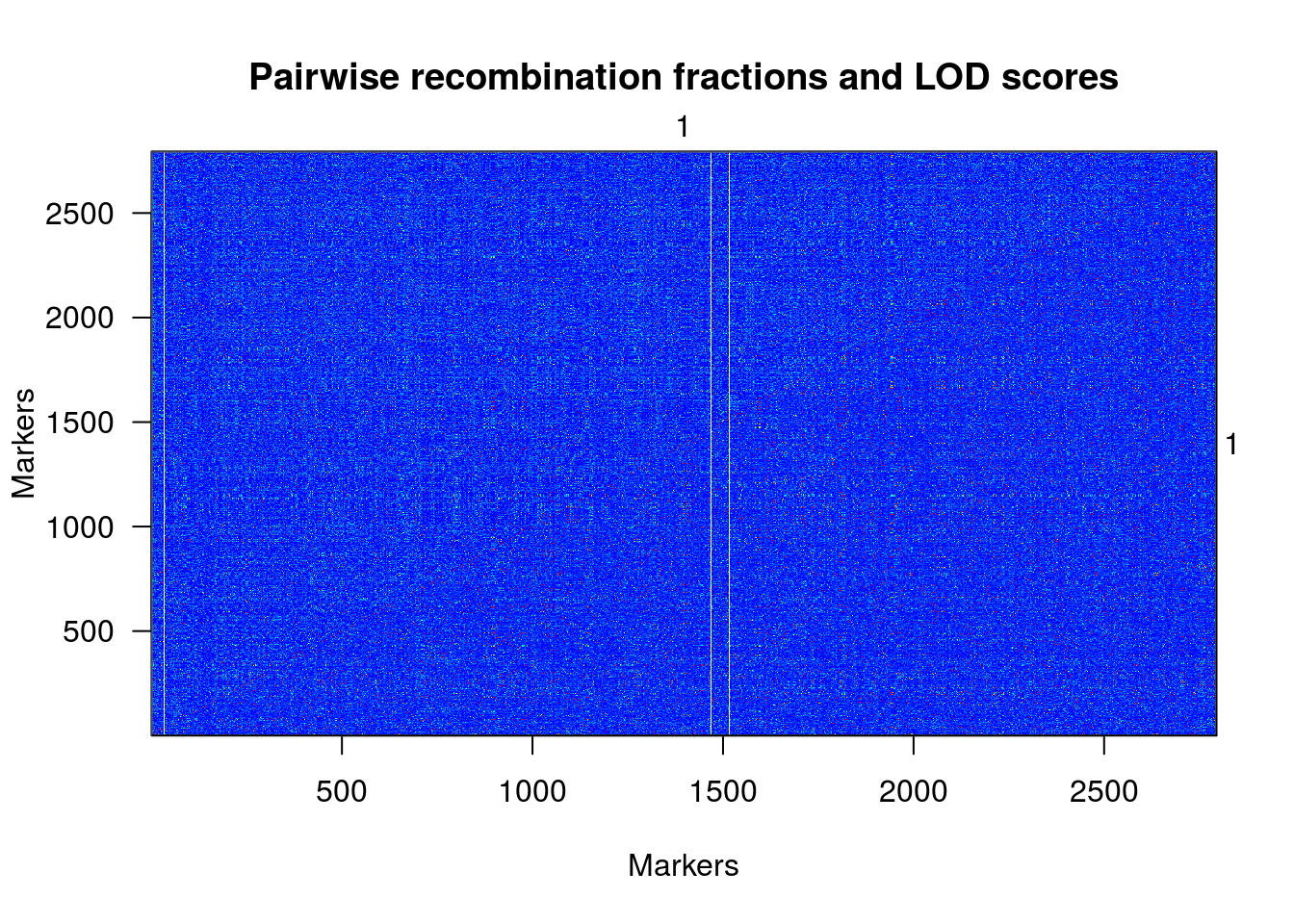
plotRF(une.cross)
## Warning in plotRF(une.cross): Running est.rf.
table(formLinkageGroups(une.cross, max.rf=0.35, min.lod=6.5)[,2])
## Warning in formLinkageGroups(une.cross, max.rf = 0.35, min.lod = 6.5):
## Running est.rf.
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13
## 472 444 333 247 201 197 182 155 155 143 137 123 5
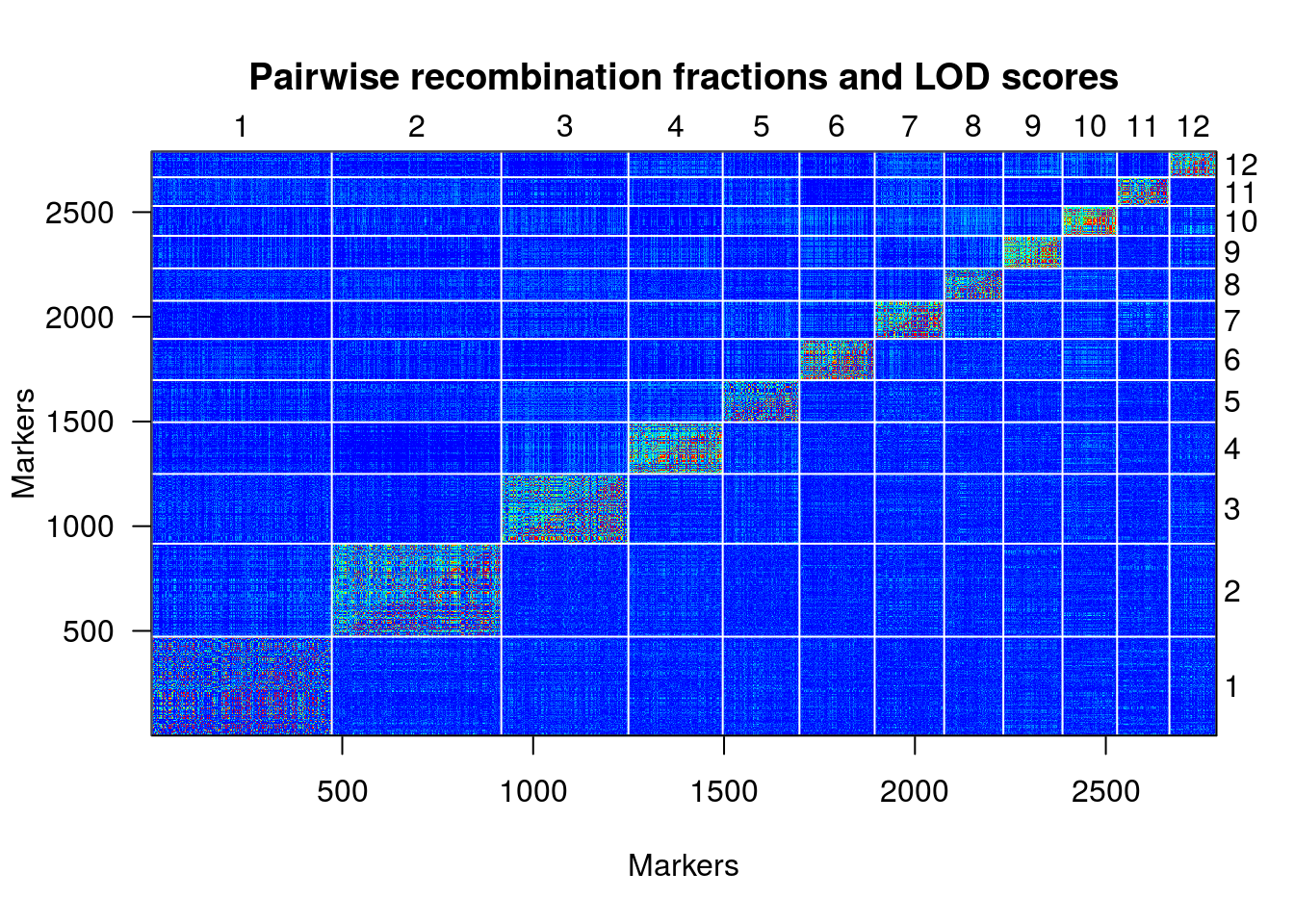
une.map <- formLinkageGroups(une.cross, max.rf=0.35, min.lod=6.5, reorgMarkers=TRUE)
## Warning in formLinkageGroups(une.cross, max.rf = 0.35, min.lod = 6.5,
## reorgMarkers = TRUE): Running est.rf.
plotRF(une.map, chr=c(1:12))
une.map <- orderMarkers(une.map, chr=c(1:12), use.ripple=F, map.function="kosambi", maxit=1000, tol=0.01, sex.sp=F)
plotRF(une.map)
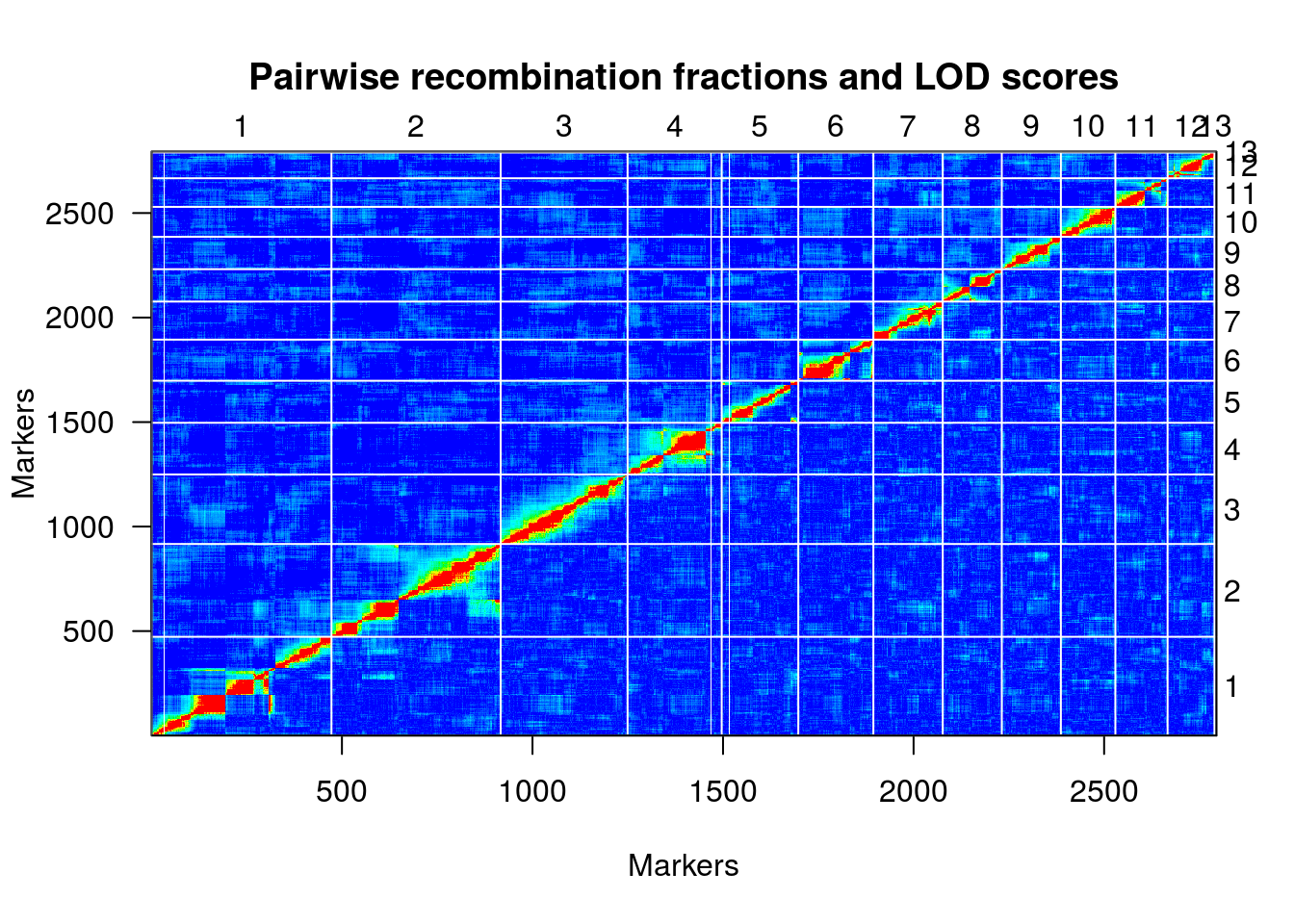
plotMap(une.map, show.marker.names=FALSE)
summaryMap(une.map)
## n.mar length ave.spacing max.spacing
## 1 472 683.8 1.5 270.5
## 2 444 398.9 0.9 134.3
## 3 333 218.3 0.7 13.7
## 4 247 237.9 1.0 30.4
## 5 201 430.5 2.2 270.5
## 6 197 231.9 1.2 57.8
## 7 182 148.3 0.8 11.3
## 8 155 185.7 1.2 55.7
## 9 155 129.4 0.8 9.3
## 10 143 105.8 0.7 5.3
## 11 137 188.2 1.4 37.9
## 12 123 151.4 1.2 27.5
## 13 5 40.0 10.0 10.0
## overall 2794 3149.9 1.1 270.5
outfile <- "uneak.om"
write.table(c("data type ri self"), paste(outfile,".raw", sep=""), append=F, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
write.table(t(c(number.individ, number.markers, 0)), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
for(i in 1:number.markers) {
write.table(t(c(paste("*", marker.names[i], sep=""), marker.genot[i,], sep="")), paste(outfile,".raw", sep=""), append=T, quote=F, sep=" ", eol="\n", na="NA", dec=".", row.names=F, col.names=F)
}
Considerações finais
É importante ressaltar que os métodos utilizados aqui requerem um bom entendimento, para, então, termos razão e certeza de aplicá-los. Basicamente, esse estudo preliminar dos métodos é fundamento para o uso de qualquer programa, algoritmo, pipeline, rotina – o que seja – sobre seus dados. Então, bons estudos!